Tutorial
- Using the search bar to retrieve binding sites
- Predicted binding sites
- Accessory ligands
- Predicted ligands
- References
Using the search bar to retrieve binding sites
Basic usage
Any text can be entered in the search bar, which results in a free text search. A query term kinase cancer, for example, is translated to the search for
the binding site entries that contain the (sub-)terms kinase and cancer anywhere in their descriptions. The white space character between the terms is
interpreted as the logical AND operator.
Advanced usage
Alternatively, a query can be made specific by providing the column name in which to perform the search, and the value for this column.
For example, pdb_id=1got retrieves exactly one protein from the database.
Valid column names are:
- pdb_id - Four letter PDB identifier, e.g., 1all
- chain_id - Protein chain identifier, e.g., A, B, ...
- rest - Binding site type [compound or cofactor]
- bs_id - Binding site number according to our druggability ranking
- organism_scientific - Scientific name of the organism from which the protein originates e.g. Homo sapiens
- protein_class - Protein class [kinase]
- disease - Disease relatedness [cancer]
- ec - Enzyme Commision number e.g., 3.4.25.1
- molecule - Name of the protein e.g., PROTEASOME SUBUNIT BETA TYPE-3
- division_id - NCBI taxonomy database division identifier [0=Bacteria, 1=Invertebrates, 2=Mammals, 3=Phages, 4=Plants and Fungi, 5=Primates, 6=Rodents, 7=Synthetic and Chimeric, 8=Unassigned, 9=Viruses, 10=Vertebrates, 11=Environmental samples]
- tax_id - NCBI taxonomy identifier, e.g., 9606
The following examples show how to properly use the query expression language in the search bar to retrieve whatever binding sites you wish from the database:
- All binding sites:
leave empty - Primary binding sites (mostly orthosteric):
bs_id=1 - Secondary binding sites (allosteric/orthosteric/other):
bs_id>1(note: our method does not reliably determine if a site is allosteric) - Compound binding sites (for substrates, agonists,...):
rest=compound - Cofactor binding sites:
rest=cofactor - Binding sites ranked No. 1 and 2:
bs_id=[1,2] - Human kinase binding sites:
organism_scientific="homo sapiens" protein_class=kinase(note the use of double quotes in search terms composed of two or more words) - Human cancer-related binding sites:
organism_scientific="homo sapiens" disease=cancer - Animal binding sites:
division_id=[1,10,2,5,6] - Bacterial binding sites:
division_id=0 - Pathogenic bacterial binding sites:
organism_scientific=["BACILLUS ANTHRACIS","BACILLUS CEREUS","BARTONELLA HENSELAE",...]
Predicted binding sites
Binding sites were predicted using the ProBiS-Dock Database method for the whole PDB. Here, we showcase a few examples of potential usage, advantages and properties of the predicted binding sites.
Binding site centroids
Binding site grid is then generated for each ligand cluster by sampling hexagonal close-packed points spaced 0.2 Å apart that fall within the radius of any of the ligands’ atoms but do not overlap with any of the protein atoms. A binding site grid thus follows the contours of the molecular surface of the biological assembly and the space occupied by the predicted ligands up to 8 Å from the protein surface. Binding site centroids are then calculated as grid points sampled at approximately 3 Å intervals. Each centroid is assigned a radius of about 4 Å, and each binding site is represented by a set of overlapping centroids with radiuses that closely follow its contours.
Primary and secondary binding sites
Primary binding sites are those with rank equal to 1 and typically correspond to a main binding site in a protein. Secondary binding sites are those with rank>1 according to our binding sites prioritization score. Ligand binding to an secondary site that is also an allosteric site can lead to a conformational change within the orthosteric binding site, thus modulating the protein’s activity.[5,6] As such, secondary sites are important in proteins as they often serve as natural control loops, such as feedback from downstream products of enzymes, while also being crucial in cell signaling. The secondary binding sites in our database can readily be used in the identification of previously unknown binding locations and subsequently the design of drugs acting on previously un-targeted binding sites, potentially resulting in drugs exhibiting novel and unique scaffolds, while still acting on the same target protein as the existing drugs that target orthosteric sites.
Accurate binding sites 3D shapes for docking
Most docking algorithms require that the search space in which the algorithm samples ligand poses, is defined prior to the start of the docking process. If this space is too large, the algorithm will require long computational times and may predict non-native poses; if the search space however, is too small, this could prevent larger potentially appropriate ligands to dock. It was determined that the optimal search space for the AutoDock Vina docking program [1] is a cube centered on the co-crystallized ligand with sides having 2.9× the radius of gyration of the currently docked ligand [2]. We show that for the human adenosine A2A receptor (PDB: 2ydo), this definition can lead to a suboptimal search space (Figure 1, orange box). Here, the cubic search space is defined as the box centered on the co-crystallized adenosine with sides 2.9× radius of gyration of a reported high-affinity antagonist (PDB: 3eml; ligand code: ZMA). It can be observed that that search space calculated using this definition does not include the fenolic oxygen of the ZMA ligand, which may result in poor docking of this compound. On the contrary, the ProBiS-Dock Database method defines this binding site using a set of centroids (Figure 4, green mesh spheres) following the shape of transposed ligands from similar binding sites, which are translated to a rectangular box shape. Here, both the adeonsine and the ZMA ligands are within the binding site search space (Figure 1, green box). This extended binding site determined by ProBiS-Dock Database should enable successful docking of larger ligands that could form additional interactions with the part of the binding site unoccupied by the co-crystallized ligands in this and other protein structures.
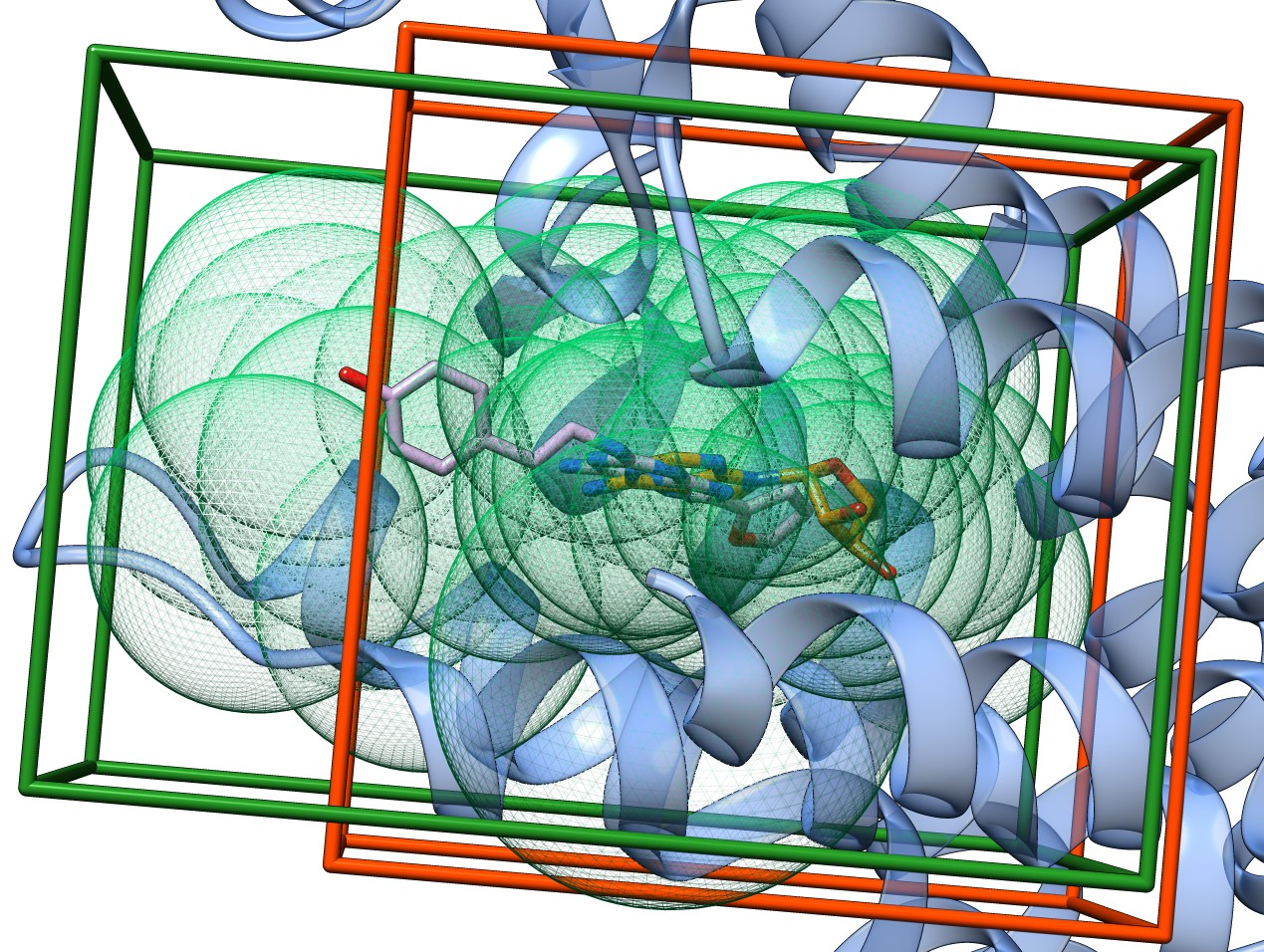
Figure 1. Alternative docking search spaces in human adenosine A2A receptor (PDB: 2ydo) (blue ribbons). The first search space (orange box) is defined by the procedure in Ref. [2] with adenosine (orange sticks) at its center. The second search space is defined by the ProBiS-Dock Database method (green box) as the smallest rectangular box that envelops the binding site centroids (green mesh spheres). The oxygen atom of the phenolic group of the ZMA ligand (pink sticks) is outside of the first search space, but within the search space defined by the green box.
Orthosteric and allosteric binding sites
Protein function can be regulated by binding of ligands to allosteric binding sites that are distinct from orthosteric sites [3]. Here, we present the ability of our method to detect allosteric binding sites in addition to orthosteric sites on the cathepsin K protein structure (PDB: 7pck). We used an apo-form of the cathepsin K structure, therefore the algorithm does not have prior knowledge about the ligands positions or binding sites. The second ranked binding site detected by our method (Figure 2, red spheres) corresponds to a known allosteric site as reported in the PDB structure 5j94, while the first ranked binding site (Figure 2, cyan spheres) corresponds to a known orthosteric site found in the structure 4dmx. The allosteric binding site is located on a flat part of the protein surface and therefore could not be detected by using, for example, cavity detection algorithms. On the contrary, in the ProBiS-Dock Database binding sites are defined based on their ligands, and thus the method can detect such sites as well.
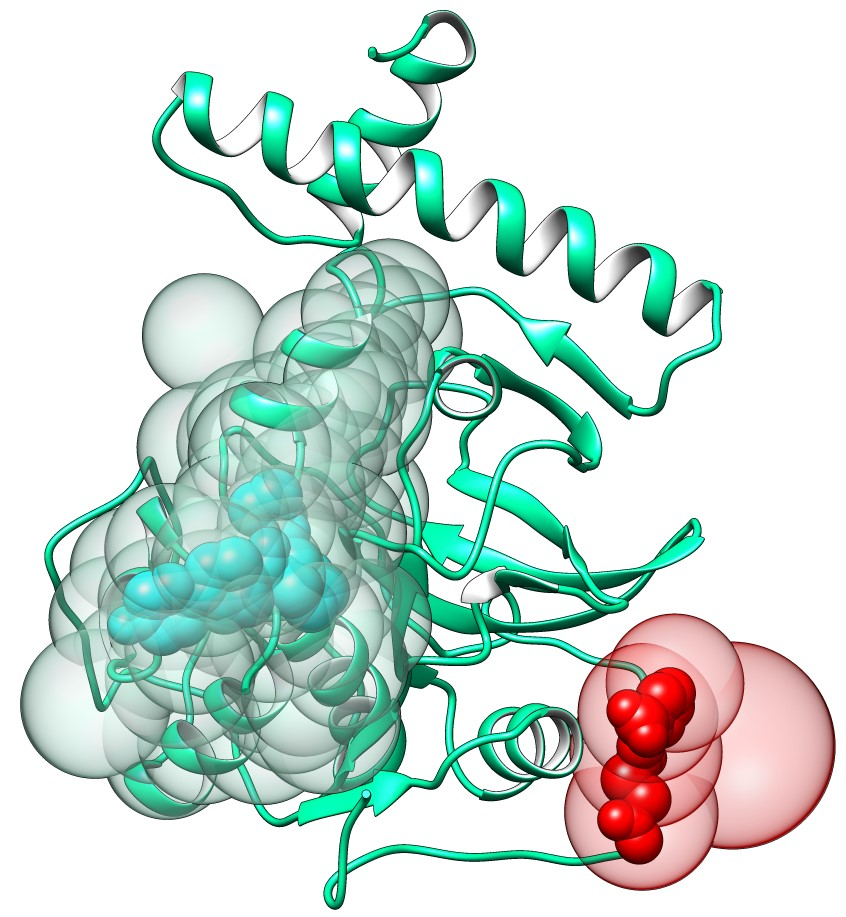
Figure 2. Allosteric binding site prediction on the structure of cathepsin K (PDB: 7pck). Allosteric binding site centroids are shown as red transparent spheres and orthosteric binding site is shown as cyan transparent spheres. Ligands within each binding site are nontransparent spheres.
Interface binding sites composed of multiple protein chains
Binding sites are often located at the interface of two or more protein chains. Using an asymmetric unit of such proteins instead of the biologically relevant assembly can lead to worse docking outcomes. Such binding sites are correctly identified in the ProBiS-Dock Database which is shown on the guanine nucleotide exchange factor (Figure 3). The ligand binding site in this protein is a cleft formed by the interface of two protein chains (A and E). With the omission of the chain E, a hydrogen bond that Tyr190 forms with the sp3-hybridized oxygen on the native ligand brefeldin A would be lost, in addition to two hydrophobic contacts with Ala157 and Thr197, which would be omitted. Taken together, considering only one protein chain would most likely reduce the accuracy of ligand docking to this binding site.
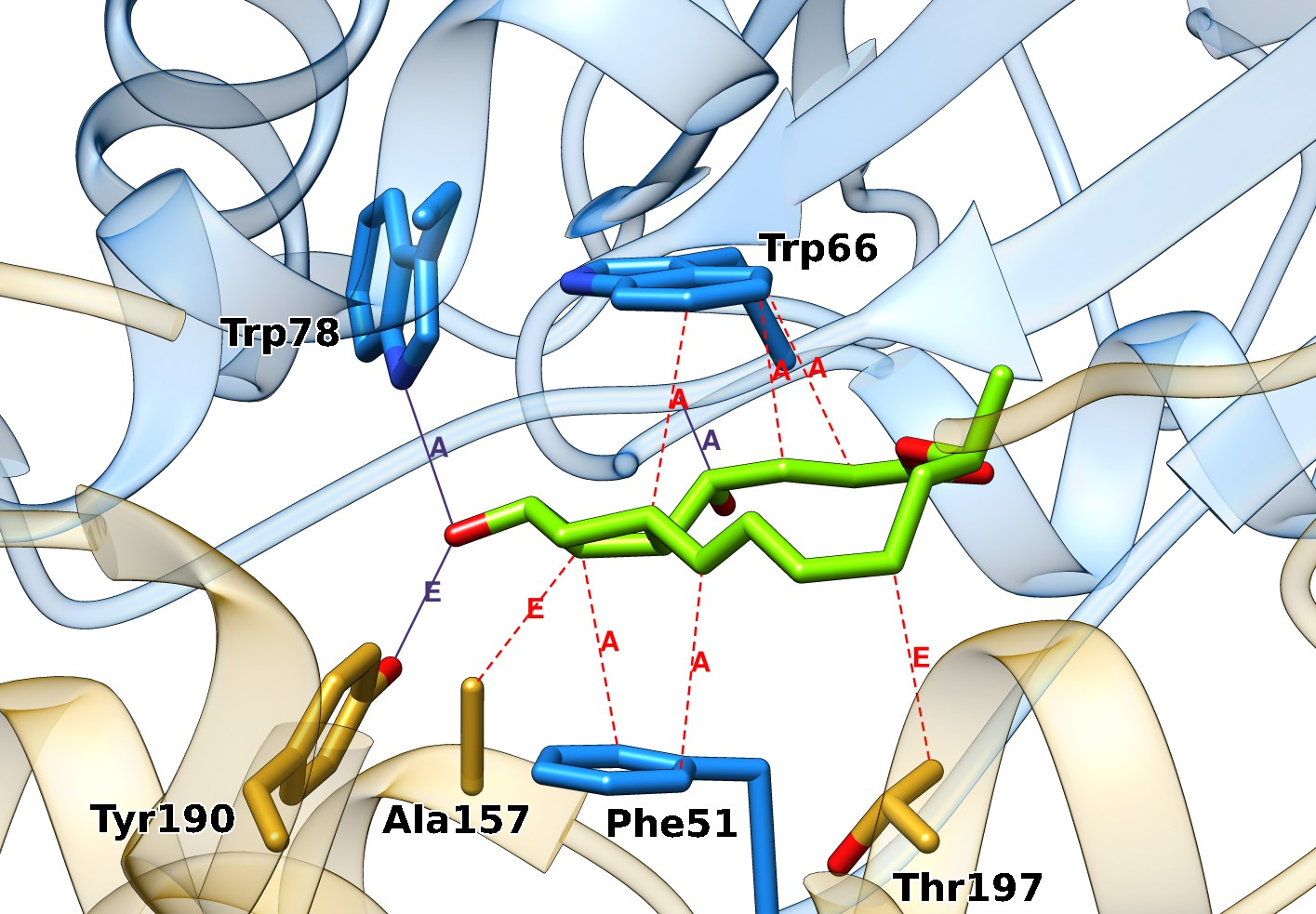
Figure 3. Interface binding site on the guanine nucleotide exchange factor (PDB: 1r8q). The ligand brefeldin A (green sticks) binds at the interface of chains A (blue ribbons and sticks) and E (gold ribbons and sticks). Hydrogen bonds are purple lines and hydrophobic interactions are red dotted lines, labeled with the chain identifiers.
Accessory ligands
Accessory ligands are ligands from the original PDB entry that may significantly influence binding of a docked ligand (eg. a drug candidate) to a protein (depending on the docking application) in the specific binding site during docking. They are determined by our method automatically. Accessory ligands are classified as cofactors, glycans, metal ions, and conserved waters.
Cofactor accessory ligands
Cofactor accessory ligands and cofactor binding sites are identified based on the list of known cofactors extended with a few more. The coordinate file for each cofactor are obtained from the PubChem database, and, basically, all PDB ligands that are very similar to the cofactors in this list, are considered cofactors themselves. The cofactors that we consider are the following:
- Adenosine-5'-diphosphate
- Adenosine monophosphate
- Guanosine-5'-triphosphate
- Phosphoadenosine phosphosulfate
- Pyrroloquinoline quinone
- Pyridoxal phosphate
- Thiamine diphosphate
- UDP-D-galactose dianion
- Coenzyme F420
- Glutathione
- Biotin
- Guanosine diphosphate mannose
- Coenzyme B
- Coenzyme M
- Tetrahydrobiopterin
- Heme B
- Molybdopterin
- Coenzyme Q10
- Vitamin K1 also Phytonadione
- N-Formylmethanofuran
- Tetrahydromethanopterin
- Cobalamine
- Triphosphopyridine nucleotide
- Adenosine-5'-triphosphate
- Cytidine-5'-triphosphate
- Methylcobalamin
- Flavin adenine dinucleotide
- Flavin mononucleotide
- Coenzyme A
- (R)-Lipoamide
- Tetrahydrofolic acid
- beta-Nicotinamide adenine dinucleotide
- S-Adenosyl methionine
- Ferroheme A also Heme A
- Vitamin C also Ascorbic acid
Glycan accessory ligands
This is the list of known monosaccharides that are used to determine if a PDB ligand is a part of a glycan.
- Pseudaminic acid
- L-Altrose
- 2-Keto-3-Deoxy-D-Mannooctanoic Acid
- D-Olivose
- N-Acetyl-D-Allosamine
- D-Ribose
- L-Idose
- D-Taluronic Acid
- D-Guluronic Acid
- N-Acetyl-D-Quinovosamine
- 6-Deoxy-L-Altose
- 6-Deoxy-D-gulose
- L-Colitose
- D-Tyvelose
- D-Paratose
- L-Apiose
- N-Acetyl-6-deoxy-L-altrosamine
- N-Acetyl-6-deoxy-D-talosamine
- Acinetaminic acid
- 4-Epilegionaminic acid
- N-Acetyl-D-Gulosamine
- N-Acetyl-L-Idosamine
- D-Xylose
- Keto-Deoxy-Nonulonic acid
- 3-Deoxy-D-Lyxo-Heptopyran-2-ularic Acid
- D-Abequose
- L-Fucose
- D-Mannose
- L-Glycero-D-Manno-Heptose
- D-Galactosamine
- L-Rhamnose
- D-Digitoxose
- D-Fructose
- N-Acetyl-D-Galactosamine
- N-Acetyl-D-Glucosamine
- L-Sorbose
- L-Arabinose
- N-Acetyl-Neuraminic Acid
- D-Glucosamine
- D-Galacturonic Acid
- D-Lyxose
- N-Acetyl-D-Mannosamine
- D-Tagatose
- D-Allose
- D-Mannuronic Acid
- D-Quinovose
- N-Glycolyl-Neuraminic acid
- D-Mannosamine
- D-Gulose
- D-Talose
- D-Psicose
- Neuraminic acid
- Muramic acid
- L-Iduronic Acid
- 6-Deoxy-D-Talose
- N-Acetyl-L-Altrosamine
- D-Glycero-D-Manno-Heptose
- D-Bacillosamine
- N-Acetyl-Muramic Acid
- L-Alturonic Acid
- N-Acetyl-D-Talosamine
- D-Glucose
- D-Galactose
- L-Altrosamine
- D-Alluronic Acid
- D-Allosamine
- Legionaminic acid
- N-Acetyl-L-Rhamnosamine
- N-Acetyl-L-Fucosamine
- N-Glycolyl-Muramic Acid
- L-Idosamine
- D-Glucuronic Acid
- D-Talosamine
- D-Gulosamine
Metal ions and conserved waters
Biologically relevant metal ions and conserved water molecules are determined by counting the number of times an ion is found in similar binding sites at the same or a similar position according to the methodology described in the ProBiS H2O approach. Biologically relevant ions are identified based on the candidate ions (see Figure 3 in the accompanying paper) and an additional filter which is used to determine that they belong to clusters of at least 10 members. Those ions that do not meet both criteria are considered artifacts and classified as buffer. Similarly, water is labeled as conserved water if it belongs to clusters with >10 members, otherwise it is considered to bind nonspecifically.
Predicted ligands
The criterion for assuming that a ligand of a protein can be transposed into a binding site on a query protein is the similarity between the binding site of the originating protein and the binding site of the query protein. Ligands are transposed from similar proteins if they have binding sites that are sufficiently similar to the binding site(s) on the query protein. Sufficiency for transposition is determined separately for each ligand type using a Z-score metric.
Prediction of ligands by transposition from similar binding sites
Z-scores are assigned by the ProBiS-ligands approach to each pairwise protein superimposition and measure the local structural similarity of the superimposed protein patches, where higher Z-scores indicate higher structural similarity of the compared binding sites.[4] For compounds, cofactors, glycans, and water molecules the superimpositions with Z-score ≥ 2.5 are used, while for metal ions this threshold is set to ≥ 2.0. Further, three different cases are distinguished for transposition: if a ligand originates from a non-representative protein within the same sequence cluster (Step 1, see our paper) as the query protein chain, then the rotational-translational matrix obtained in Step 2 is applied to the ligand’s coordinates to transpose them into the coordinate frame of the query protein chain; if the ligand originates from a representative protein of another cluster, then the rotational-translational matrix obtained in Step 3 is used; finally, if the ligand is from a non-representative protein from another sequence cluster, then both the corresponding matrices from Step 2 and Step 3 are applied to the ligand’s coordinates to transpose the ligand into the binding site of the query protein.
Nonspecific Binders
This is an updated and extended list of non-specific binders given as PDB Chemical IDs based on the list of non-specific binders available here.
12P, 144, 15P, 16D, 16P, 1BO, 1PE, 1PG, 1PS, ACA, ACE, ACN, ACT, ACY, AE3, AE4, AGC, AZI, B3P, B7G, BCN, BE7, BEN, BEQ, BEZ, BGC, BMA, BNG, BOG, BTB, BTC, BU1, BU2, BU3, C10, C15, C8E, CAC, CBM, CBX, CCN, CE1, CIT, CM, CM5, CN, CPS, CRY, CXE, CYN, CYS, D10, DDQ, DHD, DIA, DIO, DMF, DMS, DMU, DMX, DOD, DOX, DPR, DR6, DTT, DXE, DXG, EDO, EEE, EGL, EOH, EPE, ETE, ETF, FCL, FCY, FMT, FRU, GBL, GCD, GLC, GLO, GLY, GOL, GPX, HEZ, HTG, HTO, ICI, ICT, IDT, IOH, IPA, IPH, JEF, LAK, LAT, LBT, LDA, LMT, M2M, MA4, MAN, ME2, MES, MG8, MHA, MLI, MOH, MPD, MPO, MRD, MRY, MTL, MXE, N8E, NDG, NH4, NHE, NO3, O4B, OTE, P15, P33, P3G, P4C, P4G, P6G, PDO, PE3, PE4, PE5, PE7, PE8, PEG, PEU, PG0, PG4, PG5, PG6, PGE, PGF, PGO, PGQ, PGR, PIG, PIN, PO4, POL, SAL, SBT, SCN, SDS, SO4, SOR, SPD, SPK, SPM, SUC, SUL, SYL, TAR, TAU, TBU, TEP, TLA, TMA, TOE, TRE, TRS, TRT, UMQ, UNK, URE, VO4, XPE, XYP, AL, CS, BR, CL, F, IOD, PB, LI, HG, K, RB, AG, NA, SR, YT3, Y1, XE
References
- Trott, O.; Olson, A. J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31 (2), 455–461.
- Feinstein, W. P.; Brylinski, M. Calculating an Optimal Box Size for Ligand Docking and Virtual Screening against Experimental and Predicted Binding Pockets. J. Cheminf. 2015, 7 (1), 18.
- Huang, Z.; Zhu, L.; Cao, Y.; Wu, G.; Liu, X.; Chen, Y.; Wang, Q.; Shi, T.; Zhao, Y.; Wang, Y.; Li, W.; Li, Y.; Chen, H.; Chen, G.; Zhang, J. ASD: A Comprehensive Database of Allosteric Proteins and Modulators. Nucleic Acids Res. 2011, 39 (suppl_1), D663–D669.
- Konc, J.; Česnik, T.; Konc, J. T.; Penca, M.; Janežič, D. ProBiS-Database: Precalculated Binding Site Similarities and Local Pairwise Alignments of PDB Structures. J. Chem. Inf. Model. 2012, 52 (2), 604–612.
- Bu, Z.; Callaway, D. J. E. Chapter 5 - Proteins MOVE! Protein Dynamics and Long-Range Allostery in Cell Signaling. In Advances in Protein Chemistry and Structural Biology; Donev, R., Ed.; Protein Structure and Diseases; Academic Press, 2011; Vol. 83, pp 163–221.
- Kern, D.; Zuiderweg, E. R. The Role of Dynamics in Allosteric Regulation. Curr. Opin. Struct. Biol. 2003, 13 (6), 748–757.10.008.